연소성 재발성 만성염증탈수초다발신경병증 1예
Juvenile-Onset Relapsing Chronic Inflammatory Demyelinating Polyradiculoneuropathy: A Case Report
Article information

Trans Abstract
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is an acquired, immune-mediated condition. It is a symmetric, motor-predominant neuropathy that results in both proximal and distal limb muscle weakness and is characterized by electrodiagnostic or pathologic features of demyelination. CIDP is a treatable disease that is known to be rare in the pediatric population. This case report describes a 12-year-old female who presented with gait disturbance and progressive upper and lower limb weakness. The electrophysiologic findings were compatible with demyelinating polyneuropathy. Combined with the clinical features, the diagnosis of CIDP was made and treatment was administered. The first-line immunomodulatory treatment seemed to be effective, as shown by improvements in electrophysiologic and clinical parameters, but the relapsing-remitting clinical course required additional immunomodulatory treatment. Herein, we describe the patient’s clinical and electrophysiologic course according to the treatment.
Introduction
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is a progressive, symmetric and motor-predominant neuropathy that results in both proximal and distal muscle weakness in the limbs. Although CIDP was initially documented by Dyck and Tracy in 1975, its pathogenesis is still poorly understood, except about autoreactive T-cells which are known to play a dominant role in CIDP [1]. Inflammatory responses are triggered in motor and sensory nerves which cause damage to Schwann cells and myelin sheath surrounding axons [2].
CIDP is a rare disease entity with prevalence ranging from 4.8 to 8.9 cases per 100,000 persons. CIDP can occur at any age, even in early childhood and its prevalence among children is estimated to be 0.5 cases per 100,000 persons [3]. According to previous cases, juvenile-onset CIDP has more rapid onset and relapsing-remitting disease course, but more favorable long-term outcome than adult-onset CIDP. Several cases of juvenile-onset CIDP has been published in the literature, however, due to the rarity of juvenile-onset CIDP, only few cases were reported in Korea.
Although the European Federation of Neurological Societies/Peripheral Nerve Society (EFNS/PNS) consensus guideline for CIDP diagnosis is known to be most sensitive, the diagnosis of CIDP is often conflicting due to its wide spectrum of disease [4]. Intravenous immunoglobulin (IVIG), corticosteroids, and plasmapheresis are often considered as major treatment and also apply to the juvenile-onset CIDP. Juvenile CIDP shows superior response to the treatments, but a substantial proportion remains as refractory CIDP. In this case report, we described clinical and electrophysiological changes of a 12-year-old female who showed relapsing-remitting symptoms of CIDP and briefly reviewed juvenile-onset CIDP in the viewpoint of rehabilitation.
Case Report
A previously healthy 12-year-old girl was presented with a 6-month duration of progressive bilateral lower extremities weakness and gait disturbance. She did not take any medical treatment at the beginning of the symptom. However, 5 months after the symptom onset, symptom aggravated, and she started to have difficulty in walking. She was initially admitted for evaluation and treatment to the Department of Orthopedic Surgery.
Clinical examination revealed the symmetrical motor weakness of the Medical Research Council (MRC) grade 4 to 5 in the upper extremities and grade 4 in the proximal/distal lower extremities, and the Gower’s sign was positive. Hypoesthesia was examined in both medial calves. The straight leg raise test was negative, bilaterally. Diminished deep tendon reflexes were examined in the lower extremities, and there were no pathologic reflexes. Romberg test was positive and finger-to-nose test was negative, indicating no impairment of cerebellar function. There was no history of any antecedent infection and no family history of comparable symptoms. She had a history of uveitis 8 years ago. With this information, clinical impressions such as myopathy, peripheral polyneuropathy, and less likely motor neuron disease or neuromuscular disease were made.
Imaging studies, including lumbar spine magnetic resonance image, found no abnormal radiological findings. In the initial nerve conduction study (NCS) (Table 1), the compound muscle action potentials (CMAPs) showed prolonged latencies with temporal dispersion (Fig. 1), low amplitudes, and decreased conduction velocities in all the nerves tested. The sensory nerve action potentials (SNAPs) were unobtainable, with all the nerves examined. In the initial needle electromyography (EMG) (Table 1), abnormal spontaneous activities of fibrillation potentials and positive sharp waves were noted in all the right upper and lower extremities examined muscles examined. Increased proportions of polyphasic to complex polyphasic motor unit action potentials with reduced recruitment patterns were noted in the same muscles. Combined with the clinical presentation, electrophysiological findings of demyelinating disease were fitted to the CIDP criteria: (1) motor distal latency prolongation (≥ 50% above upper limit of normal) in 2 nerves, (2) reduction of motor conduction velocity (≤ 30% below lower limit of normal) in 2 nerves, and (3) abnormal temporal dispersions (> 30% duration increase between the proximal and distal negative peak CMAP) in ≥ 2 nerves. Also, the cerebrospinal fluid (CSF) protein elevation (Table 2) and negative result of PMP22 gene analysis for differential diagnosis of hereditary motor and sensory neuropathy, supported the diagnosis of CIDP. Due to the patient's young age, a nerve biopsy was not performed, but it was able to meet the diagnostic criteria for CIDP.
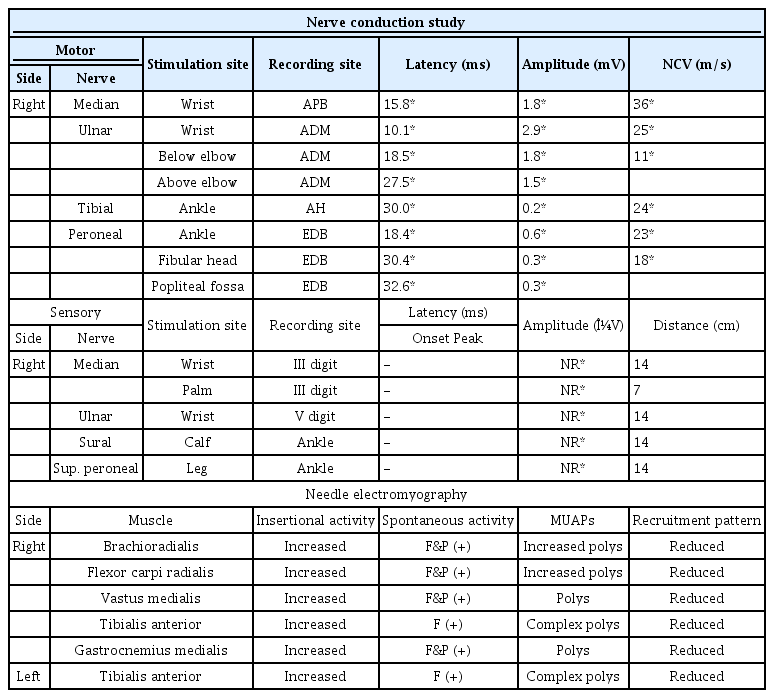
Initial (6 Months after Onset) Nerve Conduction Study and Needle Electromyography
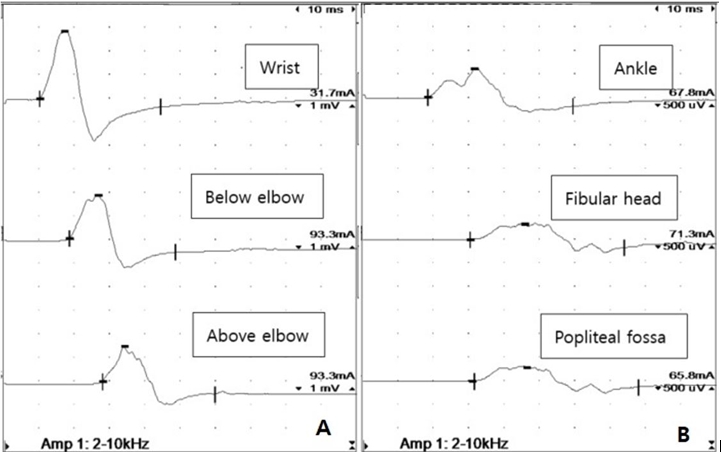
Waveform morphologies of right ulnar and common fibular nerve motor conduction studies showing prolonged latencies and prominent temporal dispersions. Amp, amplitude.

Initial (6 Months after Onset) Laboratory Findings
After the diagnosis of CIDP was made, combination of IVIG therapy (400 mg/kg/day for 5 days) and steroid pulse therapy (1,000 mg/day for 5 days) were done as the initial treatment. Also, the acyclovir therapy (10 mg/kg/dose IV3/day for 8 days) targeting varicella-zoster virus (VZV) was done. The second NCS/EMG (after 17 days from the 1st study) revealed somewhat improvement in CMAPs. She was discharged from the ward with the improvement in the gait performance, muscle power (MRC grade 5), and balance (Berg balance scale [BBS] score of 55) (Fig. 2, Table 3).

Serial distal latencies of the right ulnar nerve and timetable of serial treatments. The 5th, 7th, and 8th distal latencies of the peroneal nerve were unobtainable. The rectangular boxes with arrows denote admission events with treatment. EMG, (needle) electromyography; NCS, nerve conduction study.
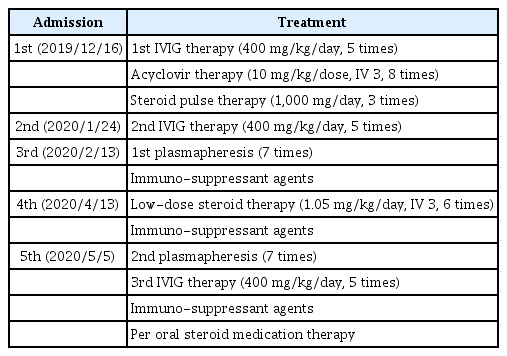
Serial Distal Latencies of the Right (Left) Ulnar Nerve and Timetable of Serial Treatments
However, she was re-admitted 3 weeks after the discharge, due to aggravation of the weakness in limbs (MRC grade 4) and gait impairment. Second-line therapy of IVIG (400 mg/kg/day for 5 days) was done and improvement of weakness was observed from MRC grade 4 to MRC grade 5. Follow-up (third) NCS/EMG after the second treatment (48 days from the 1st study) revealed continuous improvement in both CMAPs and SNAPs, but the evidence of demyelination was still noted. The SNAPs were measured in the upper extremities for the first time; onset, peak latency (ms) and amplitude (mV) were 3.7/5.0/6 and 6.8/8.9/6 in the right median (palm stimulation) and ulnar nerves, respectively. However, 2 weeks after the second discharge, the symptoms aggravated, and the patient was re-admitted. She received plasmapheresis (7 days) and immunosuppressants (azathioprine) therapy. The fourth NCS/EMG (after 99 days from the 1st study) revealed more improved results than the third one. However, the SNAPs were still unobtainable in the lower extremities. Serial NCS revealed fluctuating results in both CMAPs and SNAPs with the evidence of demyelination remaining. This pattern with relapsing and remitting symptom was continued until the last (8th) study. The motor grades of both lower limbs showed fluctuation between MRC grade 4 to 5 and the BBS score ranged from 30 to 55.
Discussion
As the patient presented with a 6-month history of progressive upper and lower extremities weakness and gait disturbance, the NCS/EMG results showed demyelinating features and met the electrodiagnostic criteria of CIDP; additionally, the clinical symptoms, high protein level of CSF analysis, and response to consequent IVIG treatment supported the diagnosis of “definite CIDP” according to the EFNS/PNS guideline of CIDP.
A previous study [5] revealed juvenile-specific clinical and electrophysiological features of CIDP compared to adults and elderly. Predominant weakness in lower extremities and deep sensation or proprioception impairment are common features of all spectrum of ages. But moderate motor weakness was noted in the juvenile-onset CIDP. Usually the decreased conduction velocity of the median nerve and prolonged distal latency of the tibial nerve is observed in electrophysiologic study of juvenile-onset CIDP, and similar results were found in our case. The motor strength of both proximal/distal lower extremities showed less fluctuation, however the gait function showed more fluctuations. This phenomenon is typical findings of CIDP since it primarily affects large myelinated fibers observed in the skin and nerve biopsy [6]. Large myelinated fibers are usually associated with proprioception, which is crucial for balancing, movement coordination, and gait function.
The differential diagnosis with hereditary motor and sensory neuropathy (e.g. Charcot-Marie-Tooth disease) is very important due to their pathophysiologic similarities. At first, CIDP presents unequal multifocal demyelination in contrast to uniform demyelination of hereditary motor and sensory neuropathy [1]. Therefore, the temporal dispersions are less-likely identified in the hereditary neuropathies. In addition to these electrophysiologic differences the disease course, CIDP shows more relapsing and remitting clinical feature compared to the hereditary neuropathy.
Conventionally, IVIG and corticosteroids are considered as first-line therapy in childhood CIDP [7]. The addition of plasmapheresis or immunosuppressant is considered if the treatment response is poor. In the previous studies of typical adult-onset CIDP, about 60% to 80% of patients were able to control disease severity with conventional therapies [1]. Children are more likely to response better to treatments. The large cohort study [7] revealed that about 80% of juvenile CIDP patients showed good responses to the first-line treatment with IVIG (79%) or corticosteroids (84%). Response to first-line plasma exchange was poor (only 14% of patients improved) although it may offer some transient or partial benefit as adjuvant or temporary therapy for selected patients [7]. However, despite this incomplete response, it is being used as an alternative treatment for patients who are ineffective with conventional treatments, such as in our case. Immunosuppressant drugs are often used for these refractory patients, and were also used in this case [6].
Guillain-Barré syndrome (GBS), which is similar to CIDP but different in onset duration, is often associated with preceding infections such as Campylobacter jejuni, Mycoplasma pneumoniae, Epstein-Barr virus, and VZV. In contrast, antecedent infection is rarely found in the CIDP. This might be because development of GBS is more influenced by environmental factors than that of CIDP. The immune system of CIDP patients are more genetically permissive, letting autoreactive T-cells to remain viable, and once activated, it causes a chronic autoimmune disease [6]. However, some previous cases reported the serologic marker of VZV found in CIDP patients. Therefore, acyclovir treatment was applied in addition to IVIG treatment in our case.
In addition to pharmacological therapy, physical therapies also benefit CIDP patients. The principal goals of rehabilitation therapy are to help the patients to achieve optimal muscle use at a tolerable pain level as nerve supply returns, and to use supportive equipment and other functional adaptations for daily activities [8]. From the acute exacerbation stage to maintaining stage, physical therapy can help the patients maintain the patients’ function and the level of daily living.
There are some proven favorable long-term prognostic factors of CIDP; subacute onset, symmetrical symptoms, good response to initial corticosteroid treatment, and predominance of nerve conduction abnormalities in the distal nerve terminals [9]. In contrast, insidious onset, asymmetric symptoms, and electrophysiological evidence of demyelination in the intermediate nerve segments are known to be associated with poor response to treatment or treatment dependent relapse. Unfortunately, this case was fitted to the latter conditions. Indeed, the onset-to-diagnosis time duration was about 6 months, which meant that the disease onset was insidious rather than subacute. The patient's mother thought her symptom as just ‘lack of fitness training’ at the early stage and sent the patient to the fitness center. Therefore, the symptoms aggravated continuously, and we can suppose that one of the leading causes for her poor disease course is late diagnosis of CIDP. There were some case reports of miss diagnosis or delayed diagnosis of CIDP, and the disease courses were poor in that case [10].
In conclusion, as in previous studies on juvenile CIDP, our patient showed remitting and relapsing clinical course and through the precise analysis of electrodiagnostic findings and physical examinations, the diagnosis and treatments of juvenile-onset CIDP were made appropriate and the medical and rehabilitative treatments were done. Juvenile CIDP is a very rare disease and appropriate differential diagnosis is crucial for suitable treatment. Future research should be considered to make more effective and additional lines of treatment to this relapsing and remitting rare disease.
Notes
Conflict of Interest
No potential conflict of interest relevant to this article was reported.